Hi greetings, sorry for asking , I had to dock NKG2A/CD94 (receptors) with HLA-E/B2M/VMAPRTLFL (ligand) proteins. And the way i did was to separate 3CDG into receptors and ligand, and the interaction based on the literature is rigid-body docking. Previously, I tried using ClusPro and used the advanced docking option of “attraction and repulsion” and since the paper only stated the interacting site , i only put attraction .
My only problem is now I am trying HADDOCK and trying to set a similar parameter with ClusPro to compare both docked models. The only problem is I am uncertain on what to do , my understanding of HADDOCK is relatively new and I think I am supposed to do an AIR docking by specifying the active/passive site of each protein. I am not supposed to do unambiguous restraint because that’s not how ClusPro computes the interaction right? I am so sorry if this post is long and doesn’t make sense, if anyone can help me , i am deeply appreciated and also will love to explain myself in details below (if needed)
Hi there,
I do not have experience with ClusPro, unfortunately, but I can clarify how HADDOCK works. To give you a very simpleidea - unambiguous restraints are useful when you know the exact distance between interacting residues (e.g. residue 5 of protein A is located at 2.1Å from residue 8 of protein B). Most often, this type of restraint is used to keep two molecules together during the docking.
On the other hand, ambiguous interaction restraints (AIRs) are used when one has general information about the binding site, something like “residue 6 of the protein A is known to be in the binding interface, but it is not clear with which residue(s) of the protein B it is interacting”.
So it seems that in your case, indeed, you need to specify the active/passive residues of each protein based on the literature. You can flip through the protein-protein HADDOCK tutorial to get a better idea of active/passive residues.
And if you don’t have any information about the interaction, you would have to turn to the ab-initio docking mode of haddock, defining for example centre-of-mass restraints.
See also our best practices guide: https://www.bonvinlab.org/software/bpg/
Hi , thanks for the reply, yes, turns out i dont have the exact distance between two interacting residues , i only have information like Residue 171 of Protein A interact with Residue 75 of Protein B, forming Hydrogen bond, but without distance, so i assume in this case , i can only do Ambiguous interaction restraints?
Also , do you mind if i asked about centre of mass restraints, surface contact restraints and when do i choose random exclusion?
Sorry for all the questions , i really am grateful for the kindness ![]()
This is then quite specific information.
You can define those two residues as active and turn off the automatic definition of passive residues.
And possibly also enable the center of mass restraints in this case.
More info about restraints here: HADDOCK2.4 manual - Ambiguous Interaction Restraints (AIRs) – Bonvin Lab
Thanks for the guidance! I’ve uploaded another trial based on these parameters! Thanks once again and hope all is well
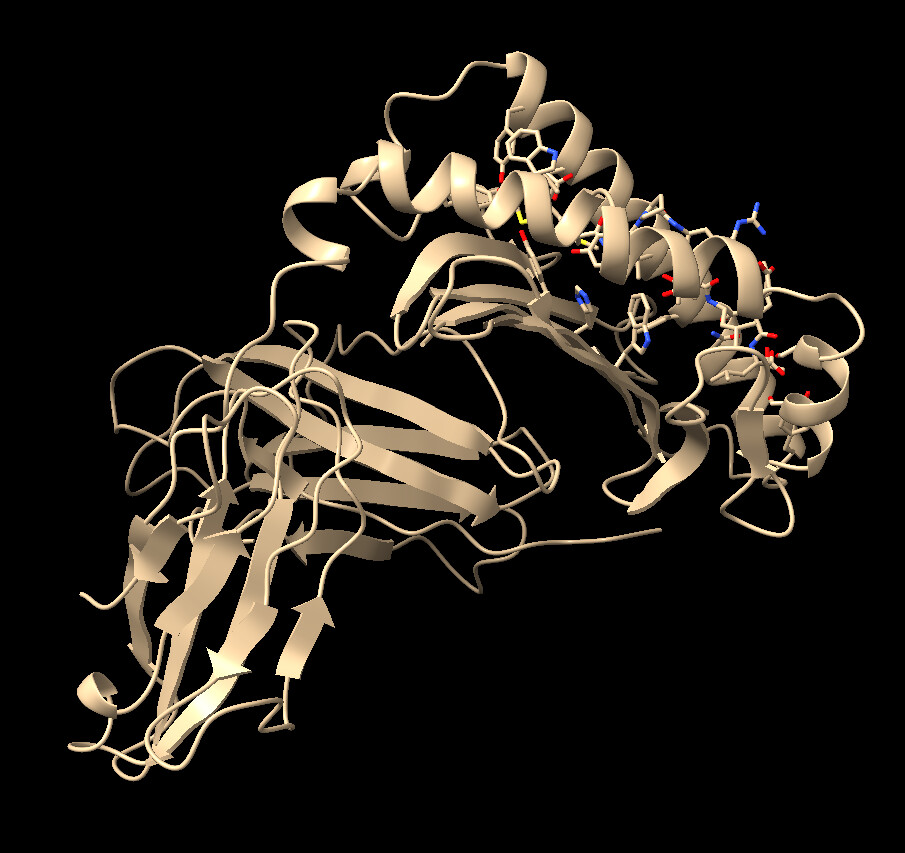
Good day, Im sorry for once again bothering everyone here , it’s just that I am still very confused. The first photo I had attached is the ligand proteins before docking, and the second photo I attached is the results of docked protein. The ligand consist of a combination of HLA-E, B2M and a nonamer peptide, all listed under a single chain B. In the second photo, I have no idea why after docking, the ligand protein has this weird linear line as highlighted in red and orange color. It appears to me that these lines exist at the border of HLA-E and B2M , along with the B2M and Peptide. Is there anything i can do to prevent this?
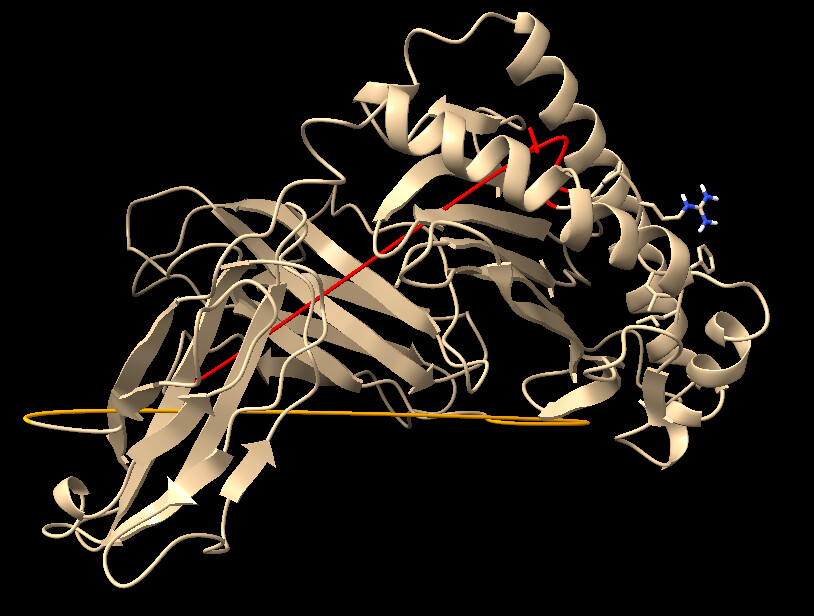
These lines are visualization artifacts - nothing to do with the model
Thanks Prof!! Relieve to hear that, dont mind if me asking why these visualization artifacts will show up ? ![]()
It depends on the software you are using.
Some might think the molecule are connected unless there is a TER statement in the PBD file.
This must be chimera(X). Try for example Pymol - it will most likely look different.