Hello PMX developers and users,
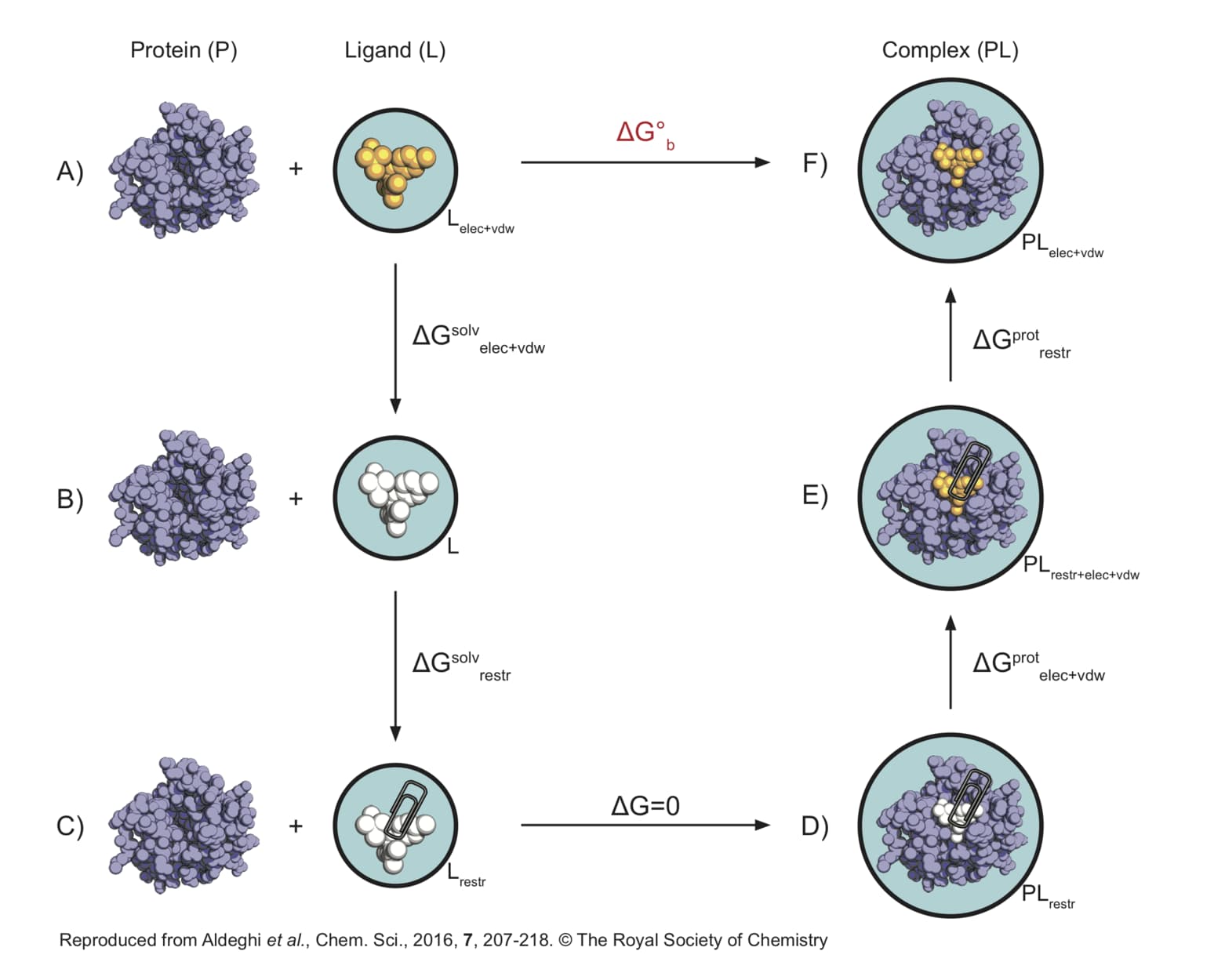
I’m setting up an absolute binding free energy (ABFE) calculation using non-equilibrium MD with PMX/GROMACS, following the thermodynamic cycle (see attached image).
- Complex Leg (D → F) in One Step?
The standard complex leg (D → F) involves two calculations: D → E (decoupling non-bonded interactions of a restrained ligand) and E → F (removing restraints from the fully interacting ligand). Is it practically feasible to combine D → E and E → F into a single non-equilibrium transformation D → F? I’m concerned about convergence, particularly for the reverse path (F → D), where a freely tumbling ligand would need to spontaneously adopt a specific restrained orientation as interactions are turned off and restraints turned on.
If it’s not possible what is your recommendation for doing that? - Boresch Restraint Selection:
For the Boresch restraints, what are the best practices for choosing the six atoms (three protein, three ligand)? Should they be on rigid substructures? Also, are there recommended GROMACS function types (e.g., [bonds] type 6, [angles] type 1, [dihedrals] type 2/9) that work best for these harmonic orientational restraints?
Any insights on these points would be greatly appreciated!
Thank you.