I am currently encountering an issue and would appreciate your assistance.
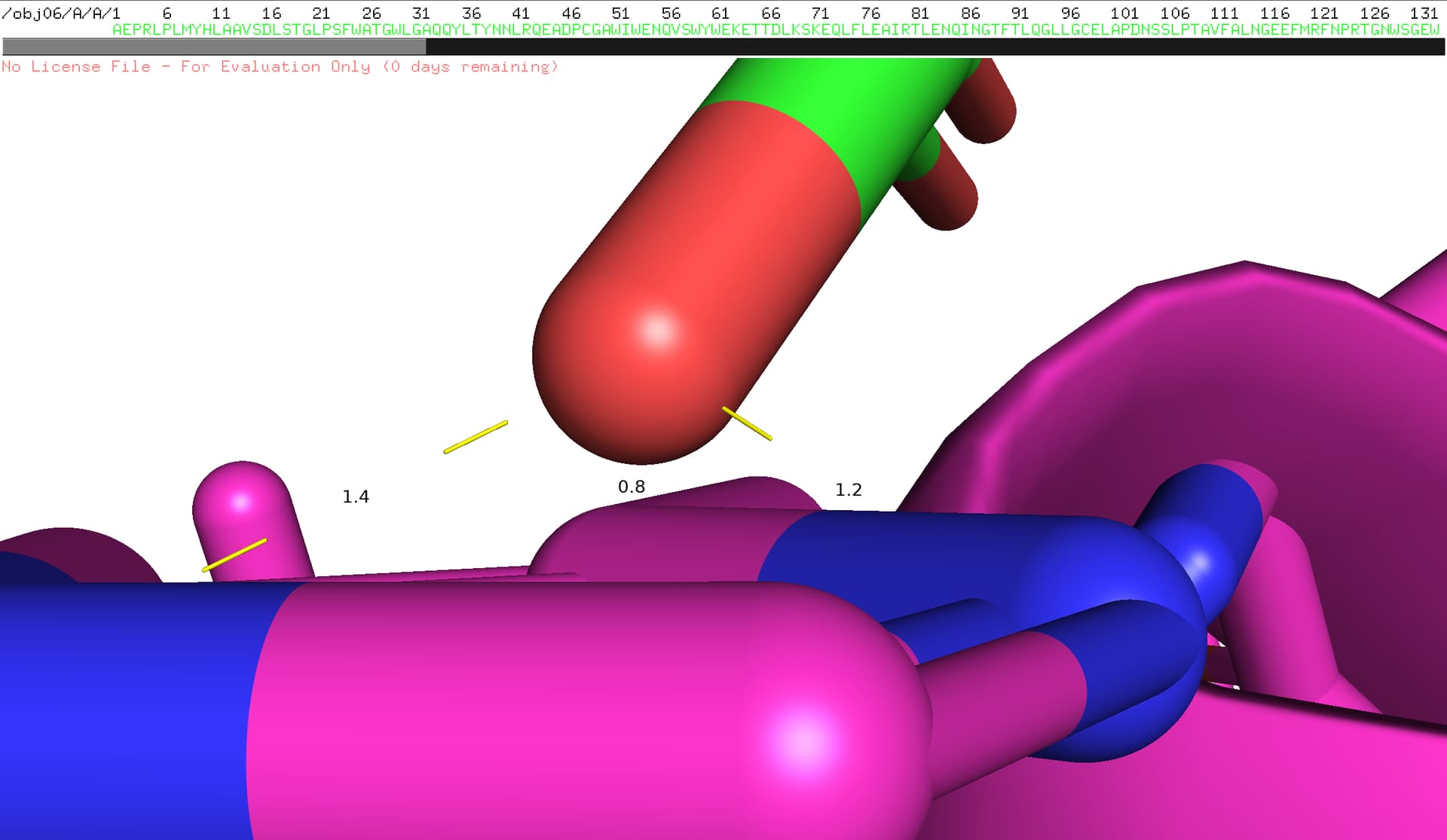
When performing docking calculations on Chains A and C of PDB ID: 1FRT using HADDOCK-score, the obtained result was: ‘HADDOCK-score (emscoring) = 136.48’. Visualization of the interface between these two chains in PyMOL revealed that certain interatomic distances are unreasonably short, exceeding acceptable steric limits (as shown in the accompanying figure).
I hypothesize that potential errors in the original structural determination of this protein complex in the PDB database may have resulted in artificially close atomic contacts between the adjacent chains. This could lead to increased steric repulsion and enhanced van der Waals forces, consequently elevating the computed score.
Could you please share your professional opinion regarding this observation?