Hello,
First of all thank you for offering this service, it is very helpful. I am trying to run an md of a ligand protein complex. The system checks the complex and even jsmol loads. However, I understand I should have the option at this point of loading parameter files for the ligand, however, a button does not appear on the screen that I can click on to get the option of loading the parameter file.
How do I overcome this problem?
Thank you for your help.
Regards,
Hi Joe,
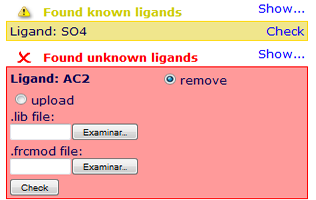
did you take a look to the ligand checking help section? ( http://mmb.irbbarcelona.org/MDWeb/help.php?id=checkingLig ). You should be able to see your ligand id in the bottom-right part of the MDWeb checking page. Just under the ligand id you should be able to find the button to upload the parameter files, as you can see in the image.
If the ligand id does not appear here, please check if its atoms in the PDB file have the HETATM tag, otherwise MDWeb will just treat them as standard residues.
Thanks for using our server.
In BioExcel we are trying to update this idea of free MD services (at least setup + configuration files) through GUIs such as web servers. Stay tuned with BioExcel web page or subscribe to our mailing list to hear about the news!
1 Like
Thank you Adam, the HETATM tag was indeed a key issue. Do you know where I can easily get the necessary parameter files for my ligand (organic molecule)?
Thank you for your help.
Regards,
Tahl Zimmerman
Hi Joe,
you can take a look at this database: http://research.bmh.manchester.ac.uk/bryce/amber/
And if you cannot find your molecule there, you can take a look at ACPYPE tool, which allows you to easily generate parameters for many different MD packages. Please note that this is an automatic generation of topology and parameters, and manual inspection is always recommended before launching a simulation with the generated parameters.
Hope it helps. Regards,
-Adam-
Thanks for all your help Adam,
I am at a new impasse. I have generated the parameter files for the ligand with ambertools leap function. I can use even use the ligand parameter files to generate files for the complex with no issues in ambertools. But when I load this ligand parameter files in mdweb I get this error:
FATAL: Atom .R.A does not have a type
repeated several times.
My ligand name is DRG however.
Do you have an idea how I can proceed?
Thanks again for you help, patience, and attention.
Regards,
Thanks for all your help Adam,
I am at a
new impasse. I have generated the parameter files for the ligand with ambertools leap function. I can use even use the ligand parameter files to generate files for the complex with no issues in ambertools. But when
I load this ligand parameter files in mdweb I get this error:
FATAL: Atom .R.A does not have a type
repeated several times.
My ligand name is DRG however.
Do you have an idea how I can proceed?
Thanks again for you help, patience, and attention.
Regards,
Hi Joe,
please could you check that the ligand parameter file (lib) corresponds to the ligand as it is in the PDB file? The name of the ligand and the number of atoms should match the ones in the PDB. The name of the atoms can be modified later on in MDWeb ligand checking section. Leap is complaining about not finding the library for the ligand.
Thanks again for using our tool!
Regards,
-Adam-
Hi Adam,
Yes the name of the ligand was indeed the problem. I had another question for you. I set up the Full Amber MD setup and I understand that at the end of all the steps it is going to carry out an MD simulation. Is this correct? For how long? I am trying to assess the stability of a docked structure. Is this MD simulation long enough to determine this?
Thank you for your help.
Regards,
Hi Adam,
Yes the name of the ligand was indeed the problem. I had another question for you. I set up the Full Amber MD setup and I understand that at the end of all the steps it is going to carry out an MD simulation. Is this correct? For how long? I am trying to assess the stability of a docked structure. Is this MD simulation long enough to determine this?
Thank you for your help.
Regards,
Hi Joe,
MDWeb was designed to help in the first steps of an MD simulation. It eases the not always straightforward setup process, and prepares the needed files to run the simulation. But unfortunately, it is not running a complete MD simulation for you. We don’t have the resources to offer that. What MDWeb offers is a short MD after the setup process (see run tutorial), with a maximum time length of 500ps. This simulation is just a test to ensure that the system has reached a stationary state, after the relaxation done in the equilibration process (included in the setup pipeline). If this equilibration was not enough for the system (it could happen with complex systems), the simulation might crash during the first steps of the “free” MD.
What you can do is follow the run tutorial, fill the parameter fields with a desired time length (for example 10ns as it is done in the tutorial), download the generated configuration files, and run the simulation in your own machine. Note that to assess the stability of a docked structure, you should probably go beyond the 100ns simulation length.
In BioExcel we are trying to update this idea of free MD services through GUIs such as web servers. Stay tuned with BioExcel web page or subscribe to our mailing list to hear about the news!
Thanks for using our tools!
-Adam-
1 Like
{kind=link}